
The FDA was forced to announce in March 2020 that it would suspend inspections of drug and medical device manufacturing sites because of the COVID-19 pandemic, but the agency has partially reversed that decision in a July 10 statement. FDA commissioner Stephen Hahn said the pause that had been applied to inspections did not prevent the agency from conducting mission-critical inspections, although routine surveillance inspections had been shuttered for four months.
The FDA has been tracking state and local conditions with the aid of a rating system designed to establish which locations are reasonably safe for a site inspection. This COVID-19 advisory rating system makes use of real-time data to provide a qualitative assessment of the number of cases in an area. These data are shared with the agency’s partners on the state level who handle some inspectional activities, presumably including inspections of mammography facilities.
The rating system is broken down into three levels of risk on a county-by-county basis, starting with the category of counties where inspections will only take place when those inspections are deemed mission-critical. The second tier of inspections takes into account whether the agency can send field investigators who have not identified themselves as vulnerable to poor outcomes for infection with the SARS-CoV-2 virus. The last tier is for counties that are open to a resumption of normal activities. The agency’s objective is to resume surveillance inspections the week of July 20, although the FDA said that conditions on the ground would continue to drive the level of inspectional activity.
FDA Posts Two Draft Updates
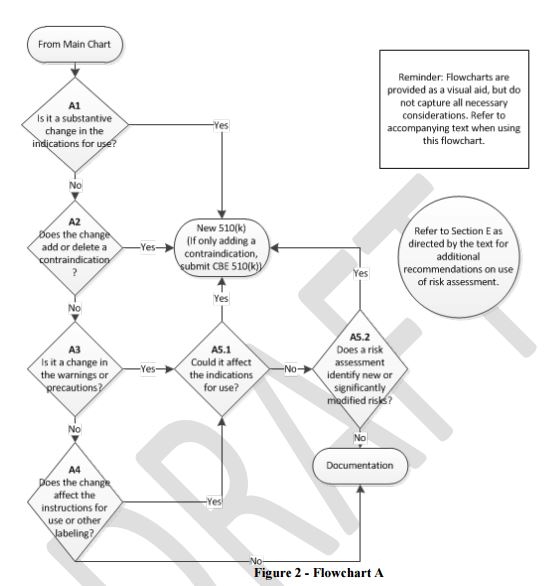
Regulatory science has not completely stagnated in the COVID-19 pandemic, a fact of life demonstrated by two draft guidances the FDA published for updates to existing guidances. One of these is a July 13 update for 510(k) submissions for devices that provide atherectomy for the peripheral vasculature, comments for which are due Sept. 11.
The scope of the draft is limited to intraluminal artery strippers that fall under the MCW product code, which includes four technologies, including rotational atherectomy devices. Sponsors can use ISO 10993 to test for biocompatibility, and ISO 14971 for risk evaluation and management. Devices that are packaged with a pre-installed internal battery will have to fulfill several performance testing recommendations, including shelf life, and the sponsor will have to determine the impact of sterilization on the battery when the battery will be left in the device during sterilization procedures.
The FDA states that it has no intention of imposing changes to the existing guidance that are not specified in this latest draft. Interestingly, the existing final guidance was issued in February 2020, less than two years after the previous draft had been published.
The second updates draft published in the month of July is the July 14 draft for clinical and non-clinical investigations into devices for treatment of benign prostatic hyperplasia (BPH), which will also limit updates to the existing to the contents of the draft. This draft will update the recommendations for devices that are covered by four product codes (KNS, PEW, PZP and NOY), and includes updates to recommended approaches to animal studies.
Among the draft recommendations is that animal studies should include both gross and histological examinations of the treated area by a pathologist who is blinded to the treatment. The draft states that animal studies of thermotherapy devices should include evaluations of how well the device limits the volume of affected tissue by checking parameters such as blood flow and tissue heterogeneity. There are also recommendations for animal studies of stents used for BPH.
Also included in the updates draft for BPH devices are recommendations for pilot and pivotal studies, including a recommendation that the sponsor make use of a randomized, controlled study design for the pivotal study. While the standard of care for the population under investigation might be the most appropriate control treatment, the FDA said the risk-benefit ratio of the investigational device should be comparable to that of the control treatment. The comment period for this draft closes Sept. 14.
 Epidemiology Network. The report described the inadequacies in the existing framework for medical device evaluation, what a more effective system would look like, what devices would be particularly ripe for evaluation via such a system, and how the system could be implemented. The full report can be found
Epidemiology Network. The report described the inadequacies in the existing framework for medical device evaluation, what a more effective system would look like, what devices would be particularly ripe for evaluation via such a system, and how the system could be implemented. The full report can be found 